2019/09 Keratin Fusion Induces Cancer Stemness by Nuclear Remodeling
Keratin Fusion Induces Cancer Stemness by Nuclear Remodeling
-
Author(s)
Jim Jinn-Chyuan Sheu Chou -
Biography
Prof. Sheu serves as a faculty member of the Institute of Biomedical Sciences, National Sun Yat-sen University. His study focuses on the discovery of cancer-driving genes and functional validation of the defined genes during cancer development. Achievements in cancer biology research resulted in Dr. Sheu winning the Academic Research Award, the Wu-Da-You Memorial Award, in 2014.
-
Academy/University/Organization
CeY3Al5O12 (YAG) and YbYAG
Cellular architecture is well maintained and organized by the cytoskeleton that provides the necessary mechanical support for various cellular functions. Three main kinds of cytoskeletal proteins have been identified in eukaryote cells, including actin filaments, microtubules, and intermediate filaments (IFs). Each type of cytoskeletal filament is characterized by a distinct type of protein subunit, filament size and intracellular distribution. Through interacting to crosslinking proteins, e.g. plectin and nesprin-3, a complex and dynamic cytoskeletal network can be established by IFs to maintain cell and tissue integrity. Loss of control of IF assembly/disassembly cycling alters the physical properties of cells in response to environment stimuli/stresses. For example, stem cell differentiation is highly mechanosensitive such that changes in IF expression profiles have been correlated with epithelial-mesenchymal transition (EMT) and cancer stem cell (CSC) formation. Such alteration can promote tumor progression, drug resistance, and metastasis through transcriptional reprogramming. The well-known ones associated with tumorigenesis include the overexpression of keratin 6 (K6), K14, K15, and K19, as well as the downregulation of K8/K18. This suggests a conserved basal epithelial program for keratin genes as a common mechanism required for the development of epithelial tumors.
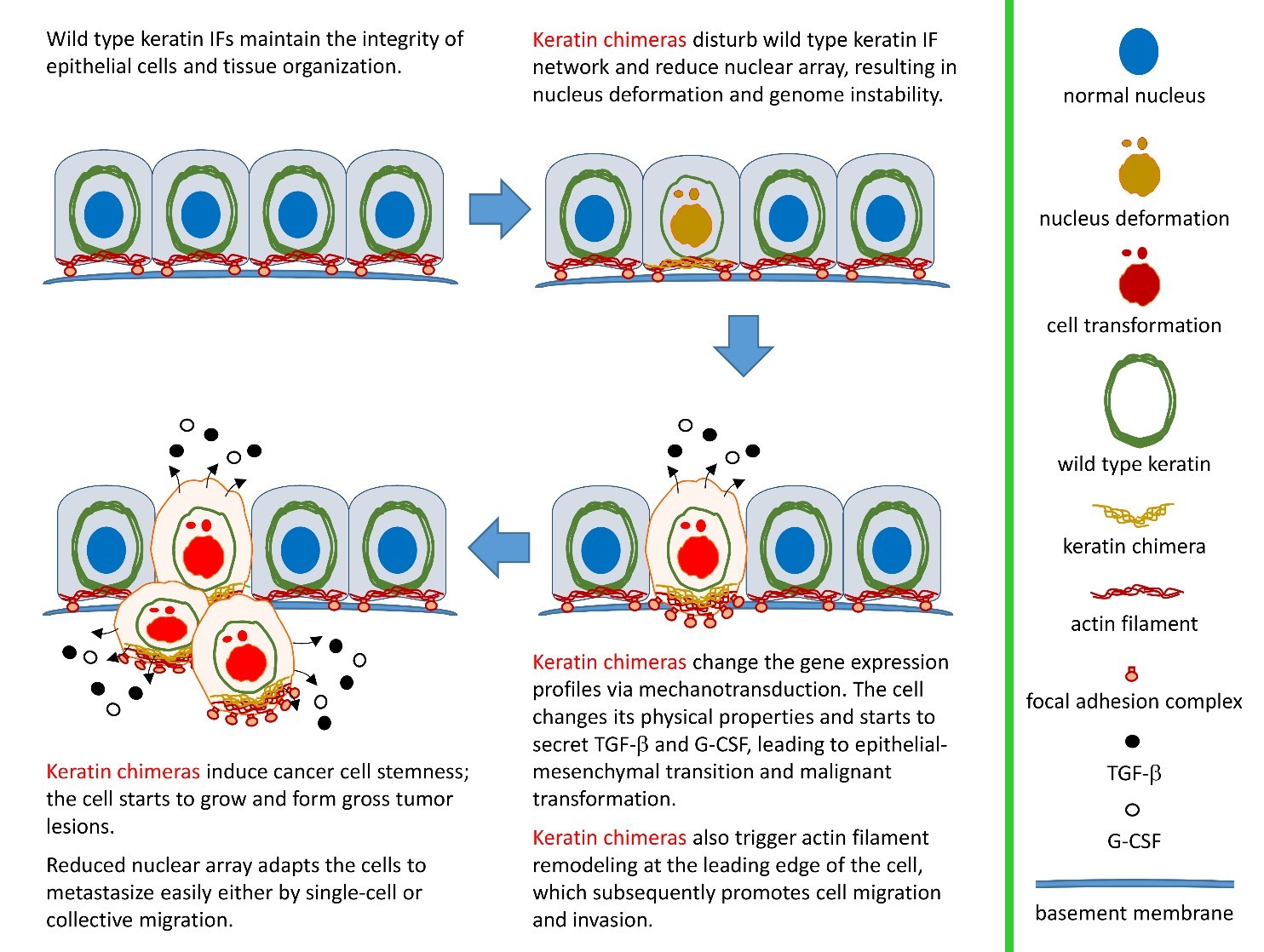
By pair-ended transcriptome sequencing, we recently found gene segments of two major basal epithelial keratins, K6 and K14, fused together to generate a variety of keratin chimeras in oral squamous cell carcinoma (OSCC). Certain types showed strong associations with carcinogen exposures and poor clinical outcomes, suggesting their potent roles in driving aggressive cancer. More importantly, such a fusion event is one of the early genetic events to trigger field cancerization because inter-keratin chimeras can also be detected in “normal-like” epithelia adjacent to OSCC, but not in blood cells collected from healthy individuals. Using a dual-fluorescent color system, we discovered that all the fusion variants (in green) showed defects in the IF assembly/disassembly cycles, and were thus unable to form perinuclear concentrated networks, although they did show interactions with wild-type K14 (in red) (Figs. 1a and 1b). Notably, loss of sufficient nuclear support made the cell nuclei deformable and fragile (Fig. 1a). DNA fragmentation and micronucleus formation were frequently detected in cells expressing keratin chimeras. Furthermore, the physical properties of those cells became much softer, which adapted them to more easily form 3-D spheres in vitro (Fig. 1c) and generated bigger and less-differentiated tumors in vivo. Since nuclear deformation is a unique feature of cell transformation, those data support our hypothesis that keratin chimeras promote tumorigenesis by interrupting the formation of wild-type keratin IF arrays in cells.
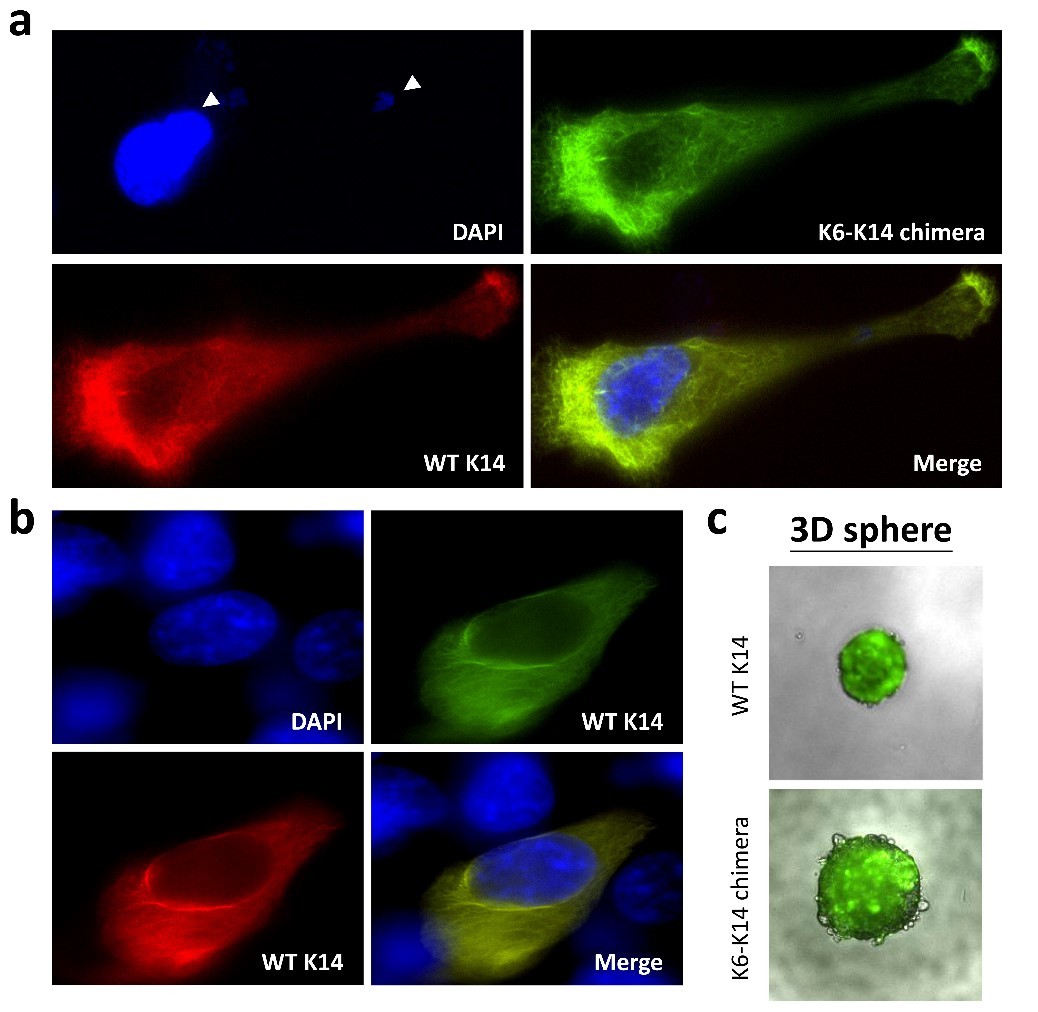
(Fig. 1)
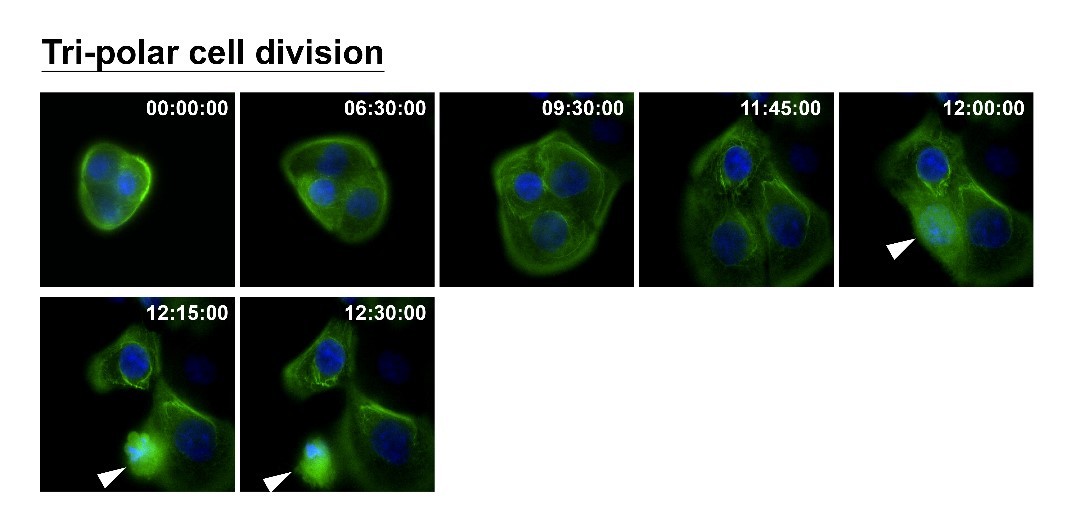
How can nuclear deformation triggered by keratin chimeras contribute to more aggressive phenotypes? It has been known that nuclear shape and position in cells are tightly controlled by a balance of cytoskeletal forces governed by IF networks. Recent studies on cell nuclei indicate a highly dynamic and coordinated concert of efforts between chromatin and the nuclear envelope in the regulation of gene expression programs. Especially, the linker of the nucleoskeleton and cytoskeleton (LINC) complex was found to play roles in connecting cytoskeletal filaments to the nucleus, performing diverse functions in nuclear positioning, mechanotransduction, and chromosome movement. In our study, we have proven the involvement of keratin chimeras in gene regulation, especially in focal adhesion and actin organization, resulting in EMT and CSC formation. Through a time-lapse study, we also found defects in chromosome segregation, leading to abnormal cell division. For example, the events of multiple-polar mitosis were significantly increased (Fig. 2), and some dividing cells tended to fuse together again after cell division, which resulted in cell death or multinucleated giant cells (Fig. 3). The keratin networks in those cells easily crashed and formed aggregates or twisted-ropes surrounding the nucleus. These findings may explain why expression of keratin chimeras can trigger pleomorphism and Mallory-body-like inclusions in mouse tumor tissues (Fig. 4), two histopathological features of aggressive cancer types.
(Fig.2)
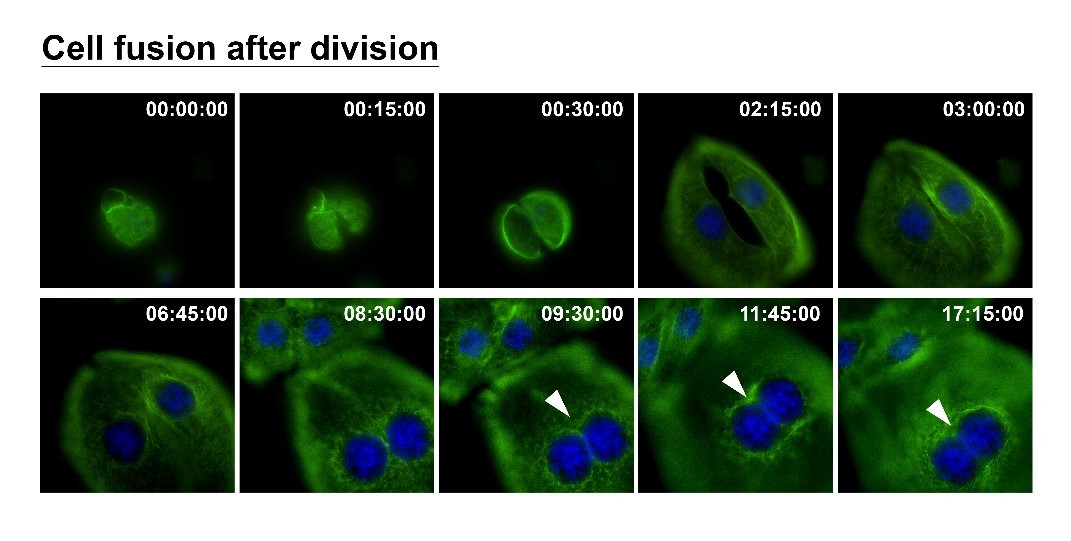
(Fig.3)
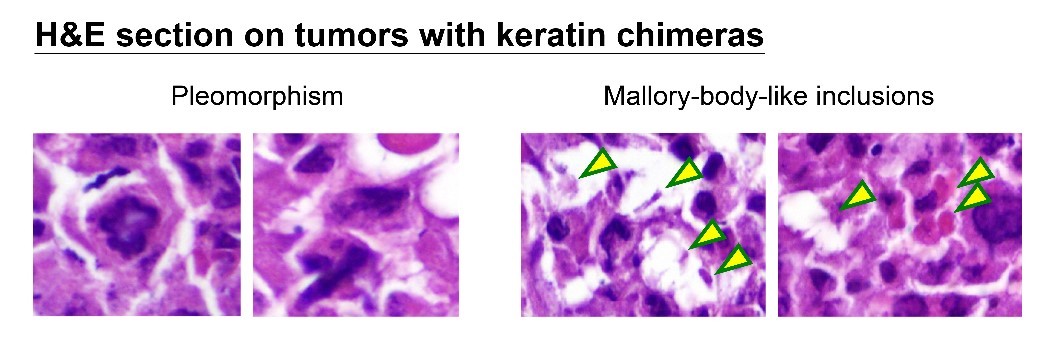
(Fig.4)
During cell progression and invasion, the cytoskeletal and nucleoskeletal framework needs to function as a whole to maintain the mechanical structures. To migrate through a complex 3-D environment, cancer cells must navigate the landscape of ECM physical properties such as pore size, stiffness and collagen types, and adapt themselves quickly to squeeze their cell shape through the matrix, requiring reorganization of the cytoskeleton. During these processes, keratin networks can provide a continuous link between ECM-cell adhesions (via focal adhesions and hemidesmosomes), cell-cell adhesions (desmosomes), and the nucleus (LINC complex). For cell clusters moving together, known as collective migration, cells also need to interact with their neighboring cells through various types of cell-cell adhesive structures. In our future study, we are going to perform quantitative biology to probe the impacts of physical properties altered by keratin chimeras on the dynamics of cellular structures during single-cell or collective migration/invasion. Since keratin fusions could also be detected in other cancer types, our data will reveal a novel molecular mechanism to link physical properties of novel IF networks established by keratin chimeras to oncogenic functionality in cancer.